In IVF practice, preimplantation genetic testing (PGT) enables the identification of chromosomal abnormalities and certain gene mutations. However, a considerable number of cases involving embryonic developmental arrest or early miscarriage cannot be fully explained by chromosomal or coding-region variations. A growing body of research suggests that such anomalies may involve dysfunctions in non-coding regulatory regions and disruptions in gene expression regulation. Nevertheless, in early human embryos, the extreme scarcity and preciousness of samples hinder comprehensive multi-omics profiling, limiting in-depth dissection of underlying regulatory mechanisms.
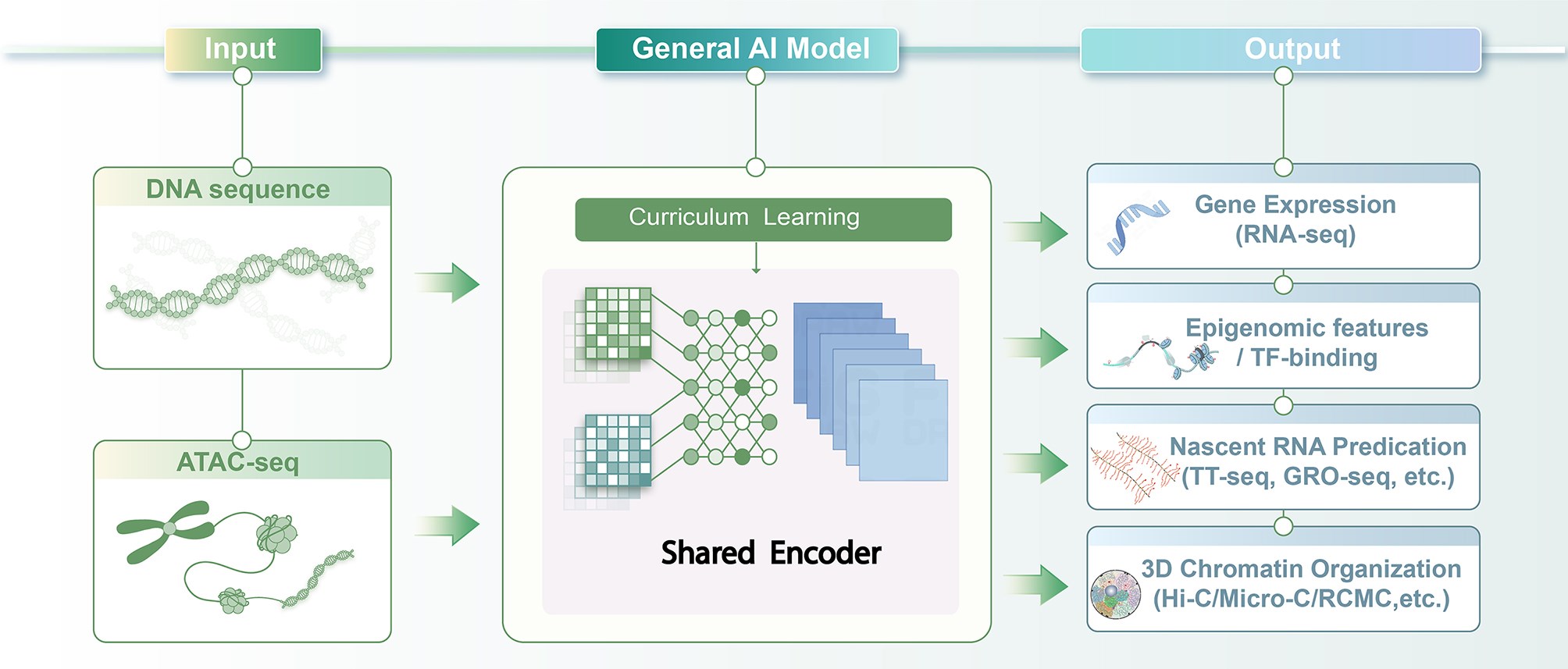
The paper presented herein introduces a multi-task artificial intelligence (AI) model that predicts multiple gene regulatory features using only DNA sequences and ATAC-seq data—a sequencing technique that maps chromatin accessibility. This work provides a theoretical foundation for integrating regulatory abnormality analysis into supplementary PGT frameworks in the future. Notably, the model still relies on chromatin accessibility input; its generalizability and resolution limits in authentic early embryonic systems require further validation. Accordingly, it is better suited as a complementary tool to experiments rather than a replacement for multi-omics assays.
Zhang Z, Bao X, Jiang L, et al. Developing a general AI model for integrating diverse genomic modalities and comprehensive genomic knowledge. Nucleic Acids Res. 2025;53(21):gkaf1269. doi:10.1093/nar/gkaf1269
Experimental Design
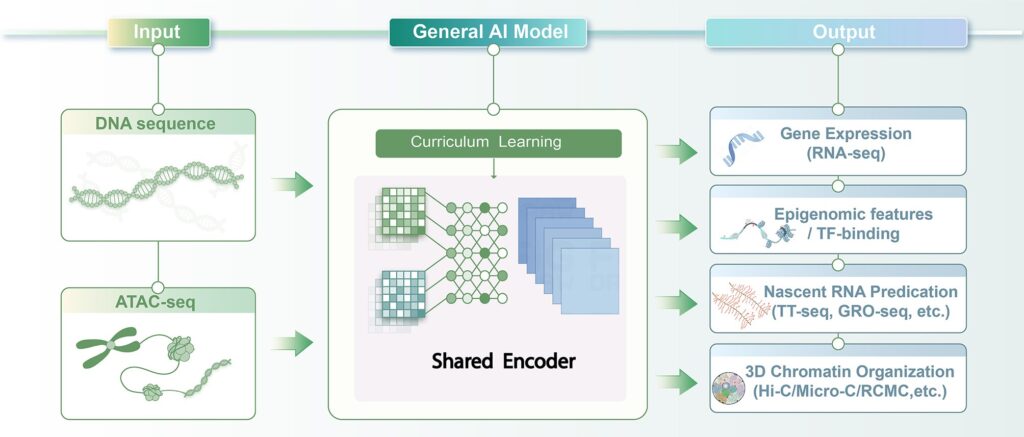
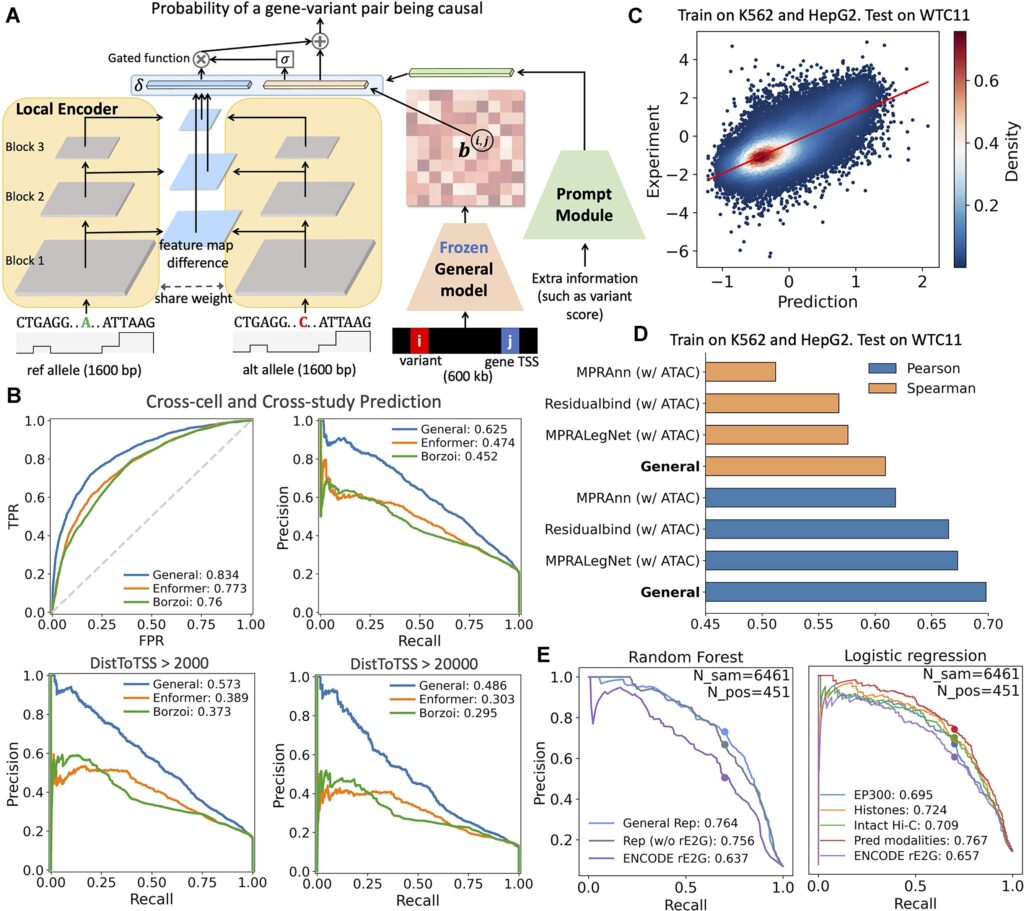
This study prioritizes versatility and data accessibility, aiming to retain predictive power for diverse regulatory features while minimizing experimental input requirements. The model only requires two types of information: DNA sequence and ATAC-seq data, which mark genomic regions with accessible chromatin. Using a unified framework, it simultaneously predicts gene expression, regulatory element activity, chromatin architecture, and other features, reducing reliance on costly multi-omics experiments.
For model training, the team adopted a phased curriculum learning strategy: the model first learns relatively straightforward expression patterns before progressing to more complex structural predictions, enhancing training stability. Additionally, the model integrates data across multiple cell types and tissues and incorporates a training scheme robust to missing data, preserving strong generalization across biological contexts.
In cross-species applications, the trained human model was transferred to the mouse genome and fine-tuned to accommodate species-specific differences, yielding a mouse-adapted universal AI model. Despite limited availability of certain mouse datasets, the model maintained high prediction accuracy, building a computational bridge for comparative and translational studies between model organisms and human development.
Key Results
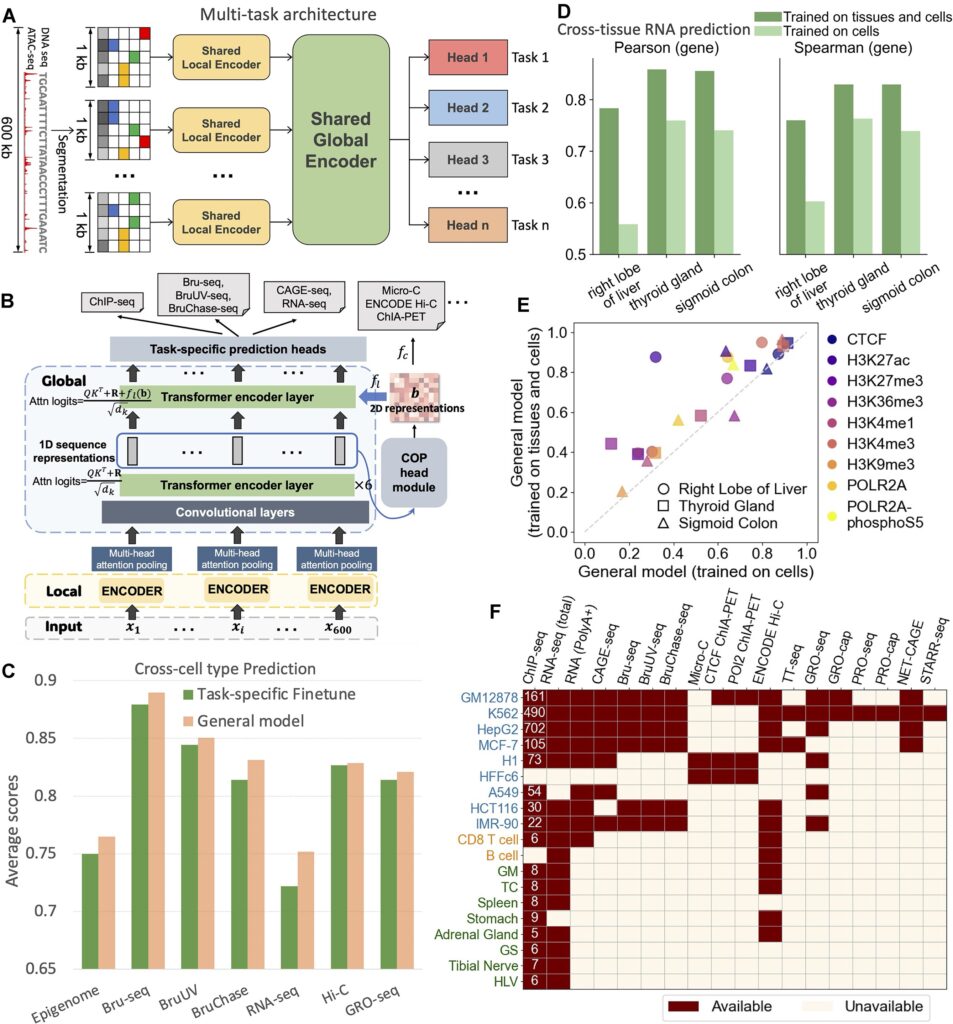
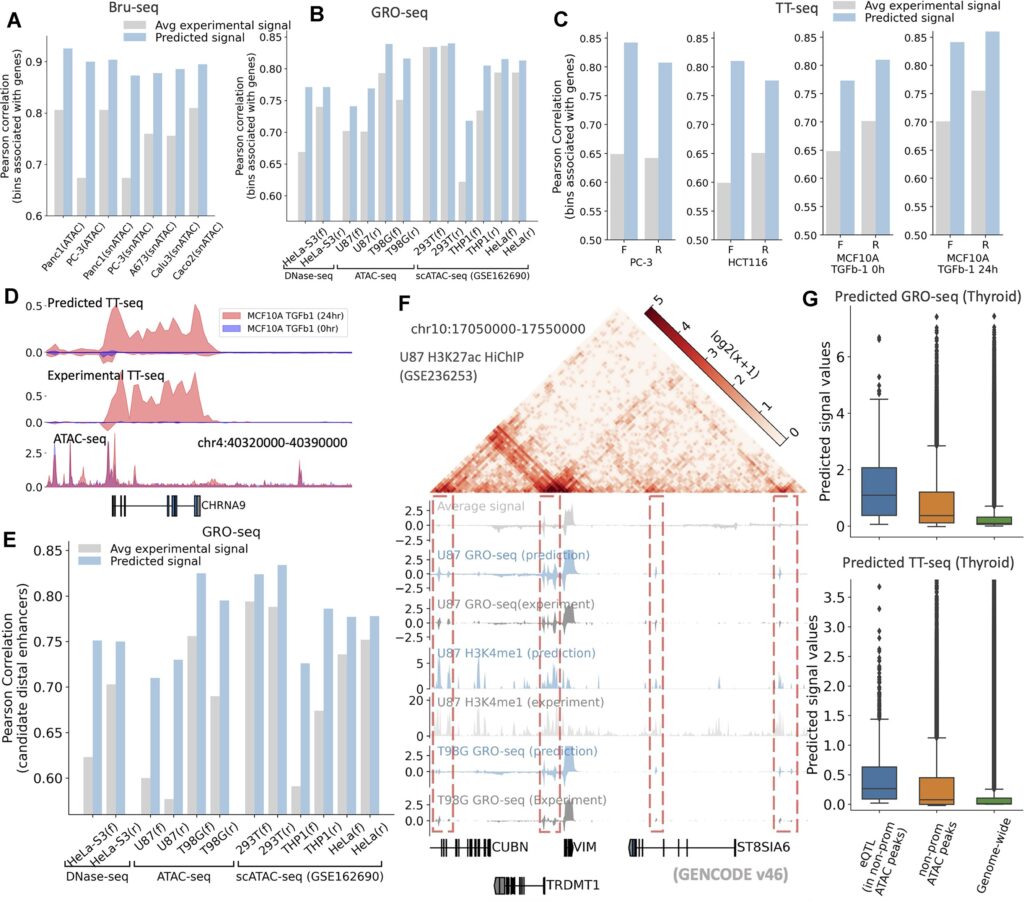
In tests on human genomic data, using only DNA sequence and ATAC-seq, the model accurately predicted gene expression levels and multiple regulatory features. For gene expression prediction, the average Spearman correlation coefficient between predicted and experimentally measured RNA-seq values exceeded 0.75. Across core tasks, performance matched or approached that of state-of-the-art single-task specialized models. The model retained high accuracy even in unseen tissue types, demonstrating meaningful generalization.
Particularly strong performance was observed in non-coding region functional prediction. When classifying expression-modulating genetic variants across 13 novel cell types, the model achieved a stable AUC above 0.82, outperforming existing mainstream prediction tools—especially in the analysis of distal regulatory regions. These results highlight its potential in identifying regulatory defects undetected by conventional chromosomal or coding-region assays.
Validation in mouse developmental models showed that gene expression profiles inferred from single-cell chromatin accessibility data achieved a Pearson correlation of up to 0.75 with experimental single-cell RNA-seq. For high-resolution chromatin 3D structure prediction, the correlation reached 0.67, confirming stable performance across species and developmental contexts.
Innovations and Limitations
The core innovation of this work lies in unifying multiple predictive tasks within one model. Instead of deploying separate models for distinct regulatory features, this single system infers multi-layer information from limited experimental data—more closely recapitulating the coordinated multi-level regulation in biological systems and better suited for sample-restricted research. The study also introduces phased training and missing-data adaptation, enabling robust performance under realistic data sparsity and imbalance. Cross-species model transfer further provides a technical route to leverage animal data for understanding human developmental mechanisms.
Several limitations remain:
- The model’s spatial resolution is relatively modest for short, finely tuned regulatory elements.
- Prediction stability for rare or developmentally unique cell types requires further validation.
- Dependence on ATAC-seq data prevents standalone application without chromatin accessibility information.
- Improvements in predicting certain chromatin contact modalities are less pronounced than in other tasks, indicating room for optimization in 3D genome modeling.
Clinical Significance and Future Perspectives
From a clinical perspective, the model’s core value is its information-completing capacity. In clinical embryo research, samples are highly precious, making multi-omics profiling on the same embryo impractical. By inferring multi-layer regulatory information from minimally invasive, readily obtainable data, the model increases information density per sample without additional biopsy or experimental burden. This facilitates mechanistic understanding of embryonic developmental competence and provides a novel analytical tool for exploring the molecular basis of developmental abnormalities.
In assisted reproductive technology (ART), current PGT focuses predominantly on numerical chromosomal abnormalities and select known mutations, while extensive regulatory information resides in non-coding regions. The model’s ability to predict non-coding regulatory functions offers a potential complementary analytical path for chromosomally normal embryos with unfavorable developmental outcomes, helping to explain regulatory defects beyond the scope of current detection methods.
For future translation into embryological research and clinical decision support, improvements are needed in resolving fine regulatory regions, expanding data coverage across developmental stages and cell types, and strengthening closed-loop integration with experimental validation. As AI and single-cell profiling continue to converge, computational prediction will play an expanding role in hypothesis generation and risk stratification. However, definitive clinical utility will require long-term, multi-center validation.